We use quantum mechanical methods to understand and predict the properties of atoms, molecules, and materials, combining concepts from quantum chemistry, chemical physics, and materials science.
1. Atomic and Molecular Physics
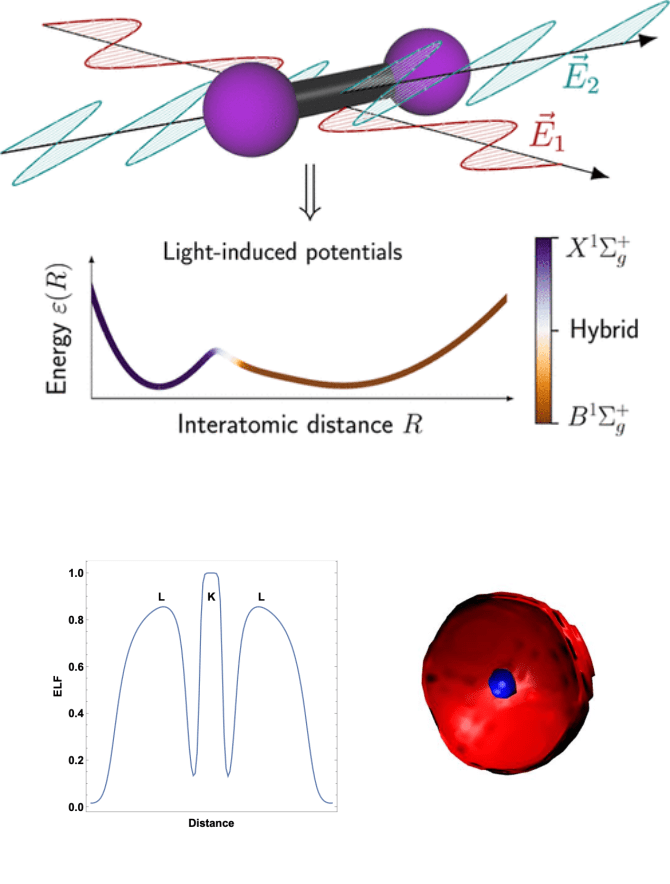
Our research in Atomic and Molecular Physics focuses on understanding how atoms and molecules behave under external perturbations such as light, confinement, or intense fields. By exploring the electronic structure of these systems, we reveal how quantum confinement and excitation affect electron localization, bonding, and reactivity.
We combine quantum-mechanical models, density functional theory (DFT), and wavefunction-based approaches to describe both ground and excited states with high accuracy. This work contributes to the fundamental understanding of matter under extreme or controlled conditions, bridging concepts from electronic structure theory, photophysics, and quantum chemistry.


2. Molecular Astrophysics
The Molecular Astrophysics line focuses on understanding molecular processes occurring in interstellar environments through quantum and quasiclassical simulations of molecular collisions.
Our work combines ab initio calculations, the development of accurate potential energy surfaces, and dynamical studies to determine reactive and inelastic rate coefficients that are essential for astrochemical and non-LTE models. This research provides fundamental insights into how molecules form, interact, and evolve under the extreme conditions of interstellar space.

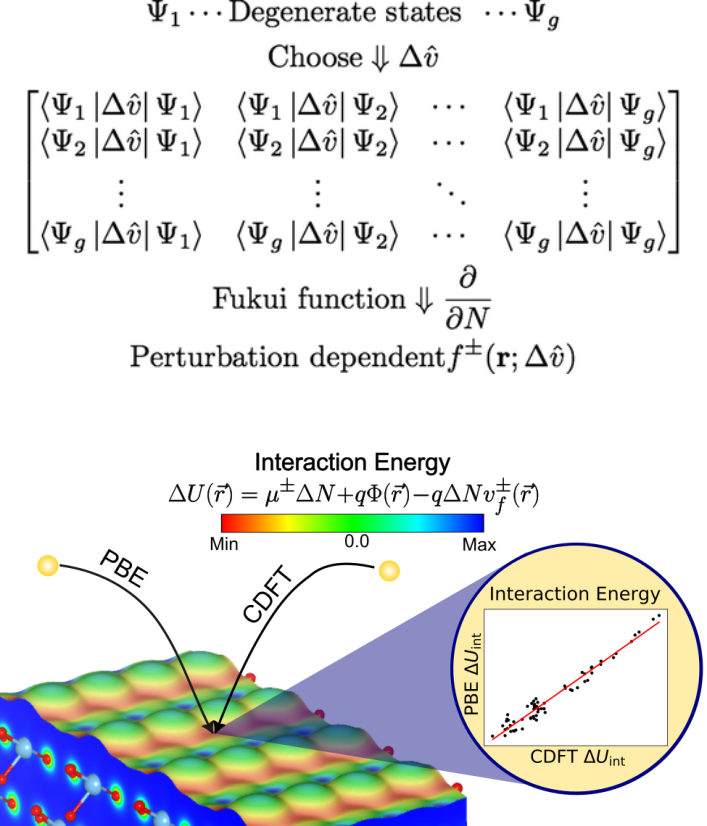
3. Chemical Reactivity Theory
Our research aims to understand and predict chemical behavior from the fundamental principles of Conceptual Density Functional Theory (c-DFT). This framework reformulates traditional chemical ideas—such as electronegativity or chemical hardness—into rigorous quantum-mechanical descriptors that link electron density to reactivity.
We have developed and applied global and local reactivity indicators, as well as new relationships among them, to identify and rationalize reactive regions in molecules, clusters, and solid surfaces. These advances have helped establish c-DFT as a quantitative framework capable of explaining site selectivity, charge transfer, and catalytic processes through density-based principles. Our approach bridges theoretical chemistry and materials science, offering a unified and predictive description of chemical reactivity across different scales.

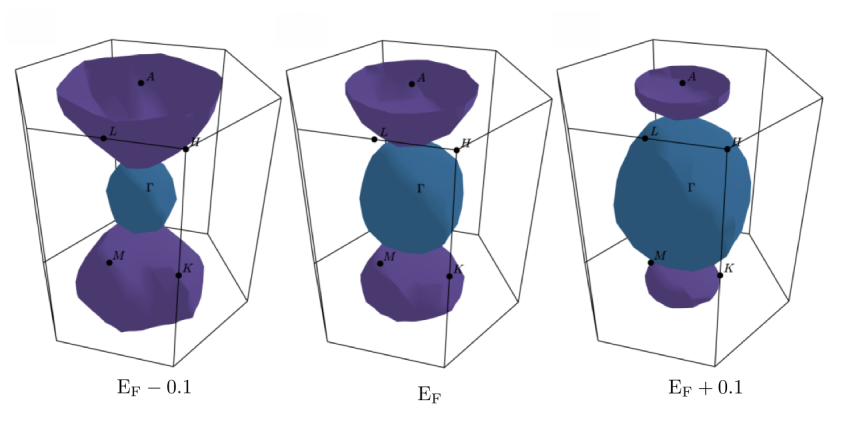
4. Material Science
Strongly Correlated System, Superconductivity, Altermagnetism, Topological Materials, Surface Reactivity
Our research in Materials Science seeks to understand how the electronic structure of materials determines their physical and chemical properties. Using ab initio and density functional theory (DFT)–based methods, we study a broad range of phenomena — from strongly correlated systems, superconductivity, and altermagnetism, to topological materials and surface reactivity.
We aim to uncover the microscopic mechanisms that connect structure, symmetry, and electronic interactions with measurable responses such as magnetism, conductivity, and photon–matter coupling. By integrating concepts from condensed matter physics and theoretical chemistry, our work bridges quantum theory and materials design, contributing to the development of next-generation functional and quantum materials.
